Химия не только изучает окружающий мир, но и активно видоизменяет его, создавая новые материалы, процессы и реакции. В этом смысле современная химическая кинетика пока делает только самые первые шаги. После того как были достигнуты определённые успехи в экспериментальном и теоретическом изучении элементарных реакций, исследователи задались вопросом: как можно управлять химической реакцией, т. е. в идеале проводить её по заранее запланированному пути и с заданной скоростью?
Для решения этой задачи надо знать различные пути реакции, т. е. иметь в своём распоряжении всю информацию о многомерной поверхности потенциальной энергии. Подобные расчёты довольно сложны даже для таких реакций, в которых участвует всего несколько атомов.
С другой стороны, химики научились произвольно изменять направление некоторых реакций. Так, используя определённую последовательность лазерных импульсов, удалось осуществить полностью контролируемую диссоциацию молекулы полутяжёлой воды HOD по любому из двух направлений:
Некоторые реакции проявляют аномальные температурные эффекты. Например, скорость биохимических реакций, катализируемых ферментами, зависит от температуры следующим образом. Вплоть до некоторой критической температуры скорость ферментативной реакции увеличивается в соответствии с уравнением Аррениуса, а затем происходит денатурация фермента (нарушение его природной конфигурации), он теряет каталитические свойства, и скорость реакции уменьшается. Это приводит, в частности, к тому, что у больных людей при высокой температуре биохимические процессы в организме протекают не так, как при нормальной.
В школьных учебниках зависимость скорости реакции от температуры описывается правилом Вант-Гоффа. Однако в подавляющем большинстве изданий по физической химии это правило даже не упоминается или же отмечается, что оно носит весьма приближённый характер и представляет чисто исторический интерес.
Дело в том, что для очень многих реакций правило Вант-Гоффа не выполняется. Так, для реакции атомов водорода с этаном Н+С2Н6=Н2+С2Н5 энергия активации Е=40,6 кДж/моль. Расчёт по формуле Аррениуса даёт ускорение в 1,69 раза при повышении температуры от 300 до 310 К (27— 37 °С) и только в 1,04 раза при повышении температуры от 1090 до 1100 К (817—827 °С), так что при высоких температурах скорость этой реакции от температуры практически не зависит. Для реакции присоединения атомов водорода к этилену Н+С2Н4=С2Н5 с низкой энергией активации (£ = 3,4 кДж/моль) скорость увеличится только в 1,8 раза при повышении температуры от 453 до 463 К (180—190 °С). Если же рассмотреть реакцию С2Н6+С2Н4=2С2Н5 с высокой энергией активации (Е=251 кДж/моль), то для неё точно такое же повышение температуры (от 180 до 190 °С) вызовет увеличение скорости уже в 4,6 раза.
Химия — наука не только экспериментальная, но и теоретическая. Можно ли теоретически рассчитать скорость реакции и предсказать скорости новых, ещё неизвестных реакций?

Константа скорости химической реакции быстро возрастает с повышением температуры.
Большинство химических и биохимических процессов с ростом температуры заметно ускоряются. Так, мясо при комнатной температуре испортится гораздо скорее, чем в холодильнике. В странах с влажным тропическим климатом фрукты созревают раньше, а машины ржавеют быстрее,
чем в северных широтах. Железо не реагирует с холодной концентрированной серной кислотой, но растворяется в горячей.
Этот эффект ещё в XIX в. был описан с помощью эмпирического (т. е. выведенного из опытных данных) правила Вант-Гоффа:
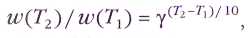
Даже по самому простому уравнению реакции нельзя сказать, является ли она элементарной или нет. Реакцию иода с водородом Н2+I2=2HI долгое время считали элементарной, потому что её скорость описывалась уравнением w = k[H2][I2], характерным для бимолекулярных реакций. Но потом выяснилось, что реакция эта сложная и состоит по меньшей мере из трёх элементарных. На первой стадии молекула иода при нагревании распадается на атомы: 12®k12I. Атомы могут либо опять превратиться в молекулу иода: 2I®k2I2, либо столкнуться с молекулой водорода и образовать две молекулы иодоводорода: 2I+Н2®k3HI
Зависимость концентрации реагента с от времени t для реакции первого порядка А®Р описывается формулой с=с0е-kt (с0 — начальная концентрация исходного вещества А, к — константа скорости реакции). Такая формула называется экспоненциальной, а соответствующая кривая — экспонентой (спадающей). Для реакции второго порядка А+®Р(в простейшем случае равенства концентраций реагентов А и В) скорость описывается уравнением w=dc/dt=-кс2 (знак «минус» показывает, что концентрация уменьшается со временем). Интегрирование этого уравнения дает иную зависимость концентрации реагента А (или В) от времени: 1/с-1/с0=kt, а соответствующая кинетическая кривая имеет гиперболическую зависимость (см. рисунок). Для реакций первого порядка период полупревращения t1/2 — величина постоянная: t1/2 =ln2/к.
Если реакции протекают при столкновении молекул, то скорость реакций должна напрямую зависеть от числа этих столкновений, которое можно рассчитать на основе молекулярно-кинетической теории. Число встреч двух частиц х и у в единицу времени прямо пропорционально произведению их концентраций: z=const [x] [у], где постоянная (const) зависит от температуры, массы и размера сталкивающихся частиц.
Встречи двух частиц длятся не более 10-12 с, частота же двойных соударений
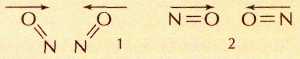
Столкновение двух молекул NO: 1 — хорошая ориентация для образования молекулы димера O=N—N=O(молекулы расположены «лицом» друг к другу); 2 — плохая ориентация («спиной» друг к другу).
Для того чтобы количественно описать зависимость скорости от концентрации, надо понять, как «устроены» химические реакции.
Они редко бывают простыми. Как правило, реакция состоит из сложной последовательности отдельных стадий. Например, окисление фосфина подкисленным раствором перманганата калия описывается ионным уравнением 5РН3+8МnО-4+19Н+=5Н2РО-4+8Мn2++12Н2О. В левой части уравнения — 32 частицы. Ясно, что все они не способны встретиться одновременно. Они реагируют друг с другом поочерёдно, объединяясь в промежуточные частицы, которые в свою очередь взаимодействуют с исходными веществами, образуя продукты реакции или другие промежуточные частицы.
Но процесс разделения сложной реакции на более простые не может продолжаться бесконечно. Есть такие реакции, которые уже не «упрощаются». Их называют элементарными.
Одна из основных задач химической кинетики -- управление скоростью реакции. Полезные реакции нужно заставить идти быстрее, а вредные — замедлить. Для этого надо знать, какие факторы влияют на их скорость.
Все химические превращения в растворе или в газовой фазе (за исключением инициируемых светом) происходят при столкновении молекул. Чем чаще молекулы встречаются, тем быстрее идёт взаимодействие. Число же столкновений, а следовательно и скорость реакции, зависит от числа молекул в единице объёма, т. е. от концентраций реагирующих веществ.
Если бы при каждом столкновении молекулы вступали во взаимодействие, все химические реакции заканчивались бы мгновенно, многие — со взрывом. На самом деле некоторые молекулы при столкновении превращаются в продукты реакции, а другие — нет. От чего это зависит?
Дело в том, что для разрыва или ослабления старых химических связей нужна энергия. Когда сталкиваются активные молекулы, которые обладают некоторым запасом энергии, они могут прореагировать. Если же энергия мала, то столкновение не приводит к реакции и молекулы разлетаются без химического превращения. Энергия молекул в свою очередь зависит от температуры. Это и есть второй важнейший фактор, определяющий скорость реакции. Подавляющее большинство реакций ускоряется с ростом температуры.
Золотые украшения сохраняют свою красоту и блеск веками. А вот брошенный на улице старый автомобиль спустя несколько лет превращается в груду ржавого металлолома; долька яблока уже через несколько часов покрывается бурой плёнкой; петарда, брошенная в костёр, оглушительно взрывается.
Любопытно, что с точки зрения химической термодинамики возможны все перечисленные процессы, даже окисление золота. Просто у них разные скорости. Одной реакции требуются для завершения микросекунды, другой — миллионы лет. Почему так? Термодинамика ответить бессильна: в этой теории не учитывается время. Скорости химических реакций изучает химическая кинетика. Более того, химическая кинетика даёт ключ к управлению реакцией.
Полученный критерий самопроизвольного протекания реакции можно преобразовать в более удобную форму, если воспользоваться ещё одной термодинамической функцией — энергией Гиббса, которая обозначается буквой G и определяется как G=Н-TS. Названа она в честь одного из основателей химической термодинамики, американского учёного Джозайи Уилларда Гиббса (1839—1903).
Преобразуем выражение DSобщ =DSсист-DHсист/T>0, умножив его на -Т. Получим:
-TDSобщ=-TDSсист+DH<0. С учётом новой функции G последнее выражение приобретает вид -TDSобщ=DGсист<0.
Теперь критерий самопроизвольности реакции и её равновесия можно выразить через изменение энергии Гиббса системы;
DG<0 — самопроизвольная реакция;
DG=0 — реакция находится в состоянии равновесия;
DG>0 — несамопроизвольная реакция (самопроизвольна обратная реакция).